Gang Chen, Ph.D.
About the PI:

Academic Experience and Training:
2021-Present: Assistant Professor (Tenure Track)
Marsico Lung Institute/Cystic Fibrosis Research Center, University of North Carolina at Chapel Hill
2014-2020: Research Associate
Marsico Lung Institute/Cystic Fibrosis Research Center, University of North Carolina at Chapel Hill
2013-2014: Postdoctoral Fellow
Cystic Fibrosis Research Center, University of North Carolina at Chapel Hill
2011-2012: Postdoctoral Fellow
Perinatal Institute, Cincinnati Children’s Hospital
2004-2010: Graduate Student (Ph.D.)
Molecular and Developmental Biology Graduate Program
Cincinnati Children’s Hospital and the University of Cincinnati
2002-2003: Research Assistant
Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
1999-2002: Graduate Student (MS)
College of Veterinary Medicine, Yangzhou University, Yangzhou, China
1995-1999: Undergraduate Student (BS)
College of Bioscience and Biotechnology, Yangzhou University, Yangzhou, China
.
.
Honors and Awards
2012-2013 American Lung Association Senior Research Training Fellowship
.
.
Research Experience:
SPDEF and FOXA3 are expressed in the airway mucus producing cells of chronic pulmonary diseases.
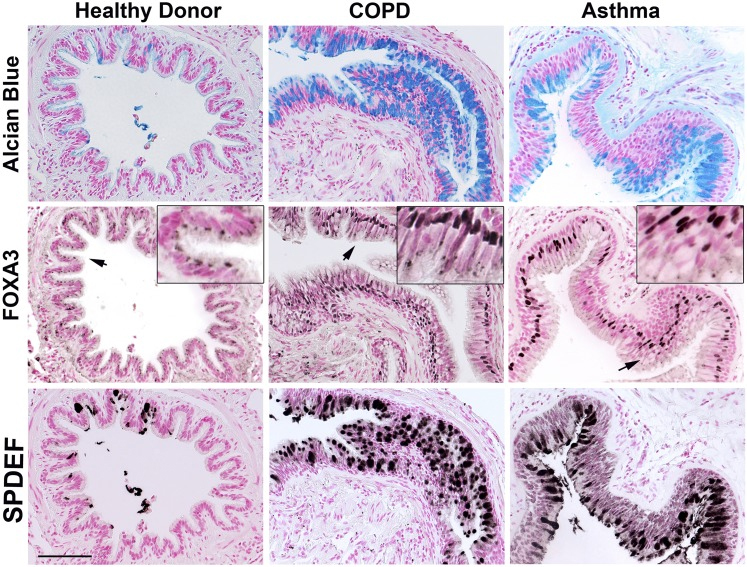
Dr. Chen was trained as a molecular and developmental biologist during his Ph.D. and as an early postdoc trained with Jeff Whitsett, MD, at Cincinnati Children’s Hospital Medical Center. Specifically, he studied the role of respiratory epithelial transcription factors in controlling lung branching morphogenesis, development, and roles of epithelial progenitor/stem cells during injury and repair. His thesis work resulted in the identification and explication of a number of important genes, and key transcription networks including SPDEF, FOXA3 (Fig.1,2), FOXA2, and NKX2.1, delineating their abilities to work as gateways that modulate MUC5AC expression and mucus production in airway goblet cells in response to Th2 dominant pulmonary inflammation that related to allergic asthma and antiviral responses in the lung.
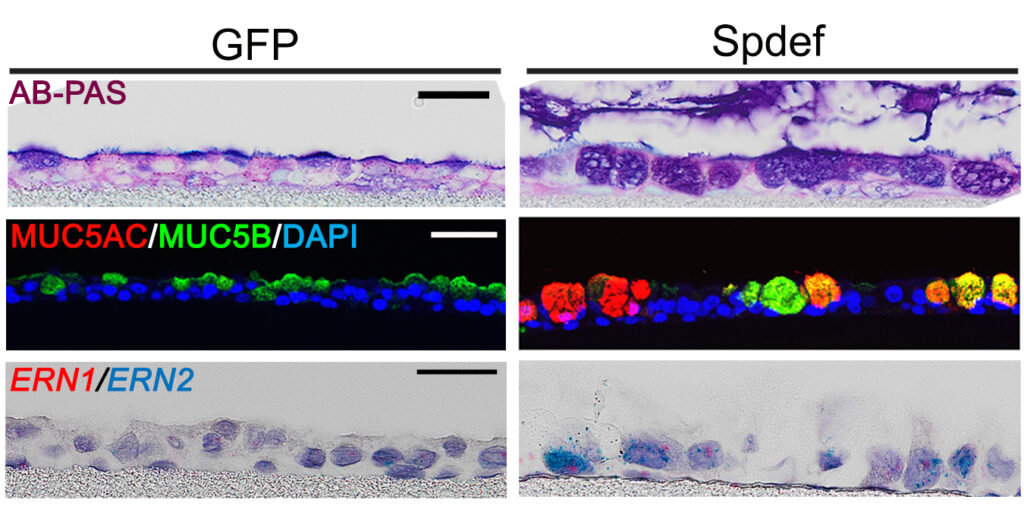
SPDEF regulates MUC5AC and MUC5B expression in primary human airway epithelial cells.
.
After Dr. Chen joined the lab of Richard Boucher, MD, in Marsico Lung Institute at UNC at Chapel Hill, he worked on discovering the transcriptional regulation of MUC5B, a major respiratory tract secretory mucins that is required at baseline for healthy, but often excessively produced in chronic muco-obstructive pulmonary diseases, like cystic fibrosis (CF), idiopathic pulmonary fibrosis (IPF), chronic bronchitis and chronic obstructive pulmonary disease (COPD).
XBP1S controls MUC5B expression in the distal airway epithelial cells in idiopathic pulmonary fibrosis lung.
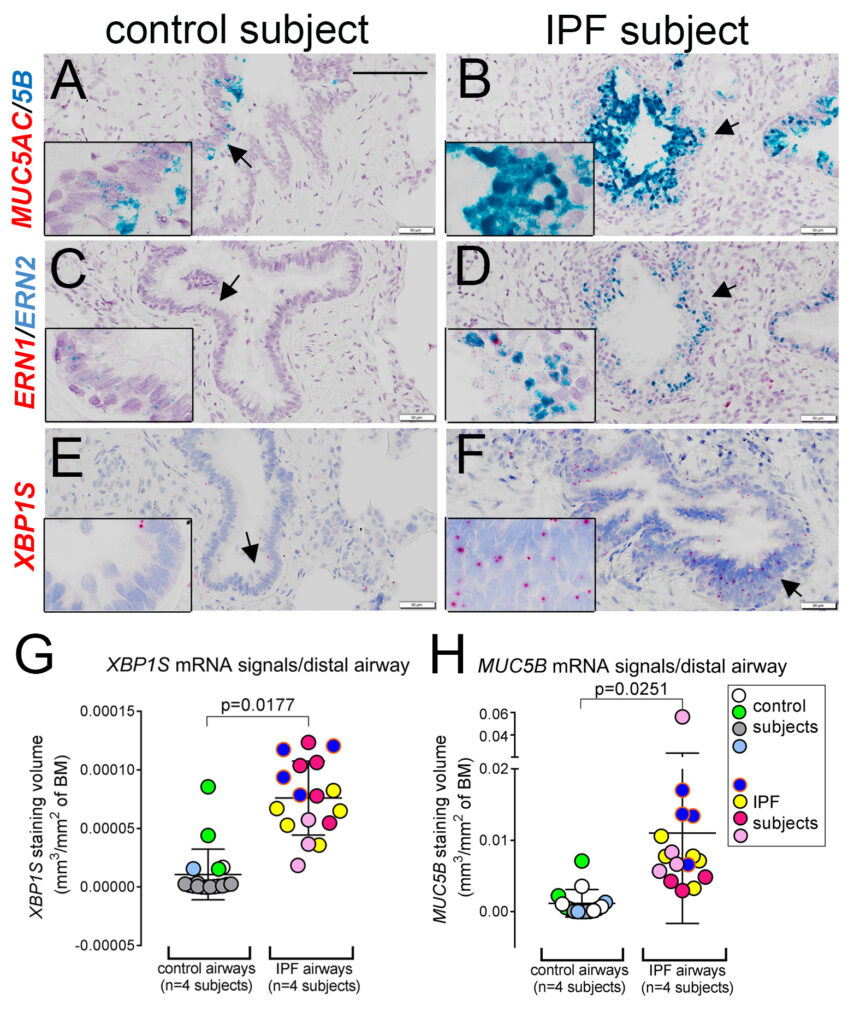
He and colleagues identified a novel pathway leading to excessive MUC5B production in IPF lung that is mediated by the SPDEF-ERN2-XBP1S axis, linking the unfolded protein response (UPR) with mucin production in the distal airway epithelia of IPF (Fig.3). Profoundly, this work presented a first-time explanation of the differential regulation of the MUC5B promoter polymorphism rs35705950, the strongest genetic association with the pathogenesis of IPF, by the ER stress transcription factor XBP1S, a potential therapeutic target.
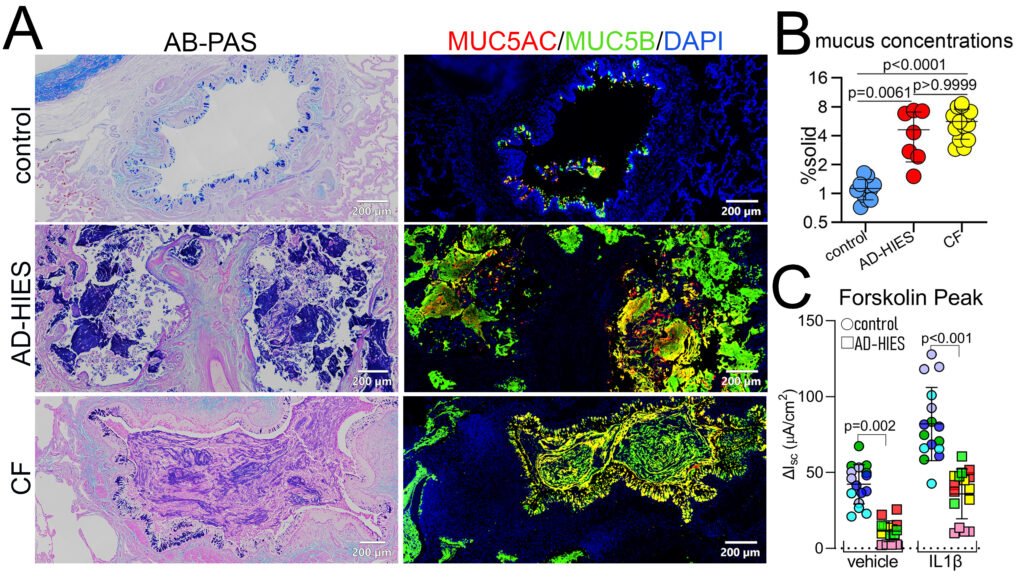
Dr. Chen also committed to studying abnormal mucus hyperconcentration and obstruction in CF. He and colleagues dissected precise transcriptional mechanisms that regulate excessive mucin production in CF airways, dominated by non-TH2 inflammation that results in hyperproduction of regional-specific mucin expression pattern, i.e. MUC5AC/MUC5B mixed in proximal vs. MUC5B dominated in distal CF airways. They identified that proinflammatory and profibrotic cytokine interleukin 1β (IL1β) is the dominant factor presented in CF airway secretions that causes abnormal mucus hyperconcentration phenotype in CF lung. They developed genetic tools to block the function of IL1β receptor (IL1R1) to suppress excessive mucin production, as well as inflammatory responses by CRISPR-Cas9 technology or by pharmacological inhibitor anakinra in primary human bronchial epithelial cells isolated from normal and CF lungs.
STAT3 deficiency leads to impaired mucociliary clearance in the airways of Job’s patients.
With strong collaborations with Drs. Richard Boucher, Kenneth Olivier at UNC, Drs. Alexandra Freeman, Kevin Fennelly at the NIH, and Dr. Peter Mogayzel at John Hopkins, Dr. Chen jointly led to study a rare genetic disease, autosomal dominant hyper-IgE syndrome (AD-HIES). This is a primarily immunological disease, due to mutations in STAT3 protein which lead to IL22/TH17 cell differentiation deficiency, and it is often associated with recurrent pulmonary infection, the leading cause of mortality in this disease. Using primary patients’ airway cells, biopsy specimens, and STAT3 CRISPR/Cas9 gene-edited normal primary cells, this team has identified the molecular and cellular mechanisms underlying the STAT3 mutations caused mucociliary clearance defects and mucus obstruction (Fig.4). These epithelial abnormalities lead to defective innate host defense and failure in the clearance of infection, that in turn lead to chronic inflammation, lung structural damage, and loss of lung function.